




By Chris A. Parr – Principal, CRO
As a follow up to “Europe, the EU, and the EU Single Market: what are the implications of geopolitics for medical device manufacturers?” we will uncover market access outside the EU/EEA based on reliance and recognition.
Regulatory reliance and recognition in countries outside of the European Union (EU) and European Economic Area (EEA) may form an important part of market access strategies. In this blog we will look at regulatory reliance and recognition based on the CE mark. We will review selected countries outside the EU/EEA in which manufacturers can leverage the CE mark as part of their regulatory strategies to achieve market access.
What is Regulatory Reliance?
Regulatory reliance, as defined in the “Draft Playbook for Medical Device Regulatory Reliance Programs” is the act whereby the regulatory authority in one jurisdiction takes into account and gives significant weight to assessments performed by another regulatory authority in reaching its own decision. The relying authority remains independent, responsible, and accountable for the decisions taken, even when it relies on the decisions, assessments, and information of others.
What is Regulatory Recognition?
Regulatory recognition (described in the same draft playbook mentioned above) is the acceptance of the regulatory decision of another regulator. Recognition should be based on evidence that the regulatory requirements of the reference regulatory authority are sufficient to meet the regulatory requirements of the relying authority. Recognition may be unilateral or mutual and may, in the latter case, be the subject of a mutual recognition agreement.
The CE Mark (Conformité Européene)
The CE mark for medical devices, while primarily intended for the EU/EEA, is also recognized or accepted in several countries outside the EU/EEA. Here is where your CE-marked medical device can potentially take you beyond the EU/EEA (map built with source material from Vemaps.com):
- Europe
- Albania
- Bosnia and Herzegovina
- Montenegro
- North Macedonia
- Serbia
- Northern Ireland
- Switzerland
- Turkey
- United Kingdom
- Asia Pacific (APAC)
- Australia
- Malaysia
- Singapore
- Middle East and Africa (MEA)
- Israel
- Saudi Arabia
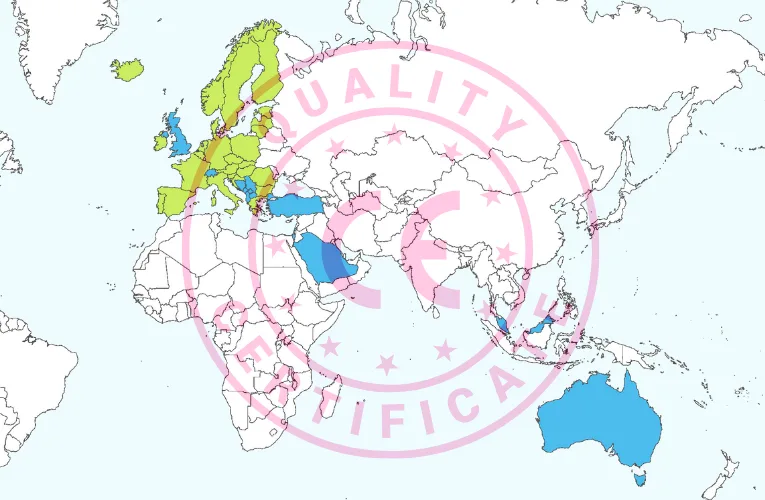
In the subsequent sections we will examine each of these countries in more detail (in alphabetical order).
Albania (Shqipëria)
Albania is located in Europe but is not part of the EU or EEA. It is aligning its medical device regulations with the MDR/IVDR and recognises the CE mark for market access. It recognises CE-marked medical devices and IVDs, meaning that if your device is CE-marked under the MDR (or IVDR), you do not need to undergo a full local conformity assessment. The CE mark is a prerequisite for registration and market access. The responsible authority is the National Agency for Medicines and Medical Devices (Agjencia Kombëtare e Barnave dhe Pajisjeve Mjekësore, AKBPM1)
Australia
In Australia, the Therapeutic Goods Administration (TGA2) allows the use of CE certificates issued under MDR/IVDR to support applications for device approval. This is part of a regulatory reliance strategy to streamline access to safe and effective medical devices. If a device has already been assessed by a notified body, then the manufacturer may be eligible for an abridged conformity assessment. This means submitting fewer documents, the TGA may reduce the depth of its own assessment, manufacturers may benefit from reduced assessment fees.
Bosnia and Herzegovina (BiH)
BiH is located in Europe but is not part of the EU or EEA. It is aligning its regulatory framework with the MDR/IVDR and recognises the CE mark for market access. However, local registration is required before a device can be placed on the market. The competent authority is the Agency for Medicinal Products and Medical Devices of Bosnia and Herzegovina (ALMBiH3). Registration is mandatory for all devices before they are placed on the market. Devices must be CE-marked under the MDR or IVDR for IVDs as applicable for the device’s classification.
Israel
Israel is located outside Europe and is not part of the EU or EEA. In Israel, the use of overseas market authorisation evidence plays a significant role in the medical device and IVD registration process, particularly under the Fast-Track Route managed by the Ministry of Health’s AMAR (Medical Devices Division4). Israel allows medical devices that have been approved in certain reference countries (including the EU) to be registered more quickly through the Fast-Track Route. Registrations via Fast-Track are valid for up to 5 years (3 years for implants). The Israeli approval cannot exceed the validity of the reference country’s authorisation.
Malaysia
In Malaysia, the CE mark is not automatically accepted for general market access, but it plays a significant role in simplifying the conformity assessment process, especially for medical devices and IVDs. Medical devices and IVDs approved by recognized foreign regulatory authorities (including CE-marked devices) can undergo a streamlined conformity assessment. Malaysia’s Medical Device Authority (MDA5) implemented Circular Letter No. 1/2025, which introduces a process called “verification”, where CE-marked devices can be registered more efficiently.
Montenegro
Montenegro is located in Europe but is not part of the EU or EEA. In Montenegro, overseas market authorisation evidence particularly from the EU and other recognized jurisdictions plays a supportive role in the medical device registration process. While Montenegro does not have a formal regulatory reliance pathway, it accepts and requires several documents that are typically issued by overseas authorities. A CE certificate under MDR or IVDR can be used to demonstrate compliance with international standards and support the local conformity assessment process with the Institut za lijekove i medicinska sredstva Crne Gore (CINMED6).
Northern Ireland
In Northern Ireland, the regulation of medical devices and IVDs is unique due to the Windsor Framework agreement which maintains alignment with EU laws for goods. Northern Ireland continues to follow the MDR and IVDR. This means that CE-marked devices under MDR/IVDR are required and accepted for placing products on the Northern Ireland market. The MHRA remains the competent authority for medical devices in Northern Ireland. However, EU Notified Bodies are responsible for conformity assessments under MDR/IVDR.
North Macedonia
North Macedonia is located in Europe but is not part of the EU or EEA. In North Macedonia, the regulation of medical devices is governed by the Agency for Medicines and Medical Devices (MALMED7) and is currently undergoing reforms to align more closely with EU standards as part of the country’s EU accession process. While North Macedonia does not have a formal regulatory reliance framework like Australia or Israel, it does accept and consider overseas market authorisation evidence, particularly from the EU and other regulatory authorities, as part of its device registration process. The CE certificate may be used to streamline the evaluation process, especially for devices already approved in the EU or other recognized jurisdictions.
Saudi Arabia
In Saudi Arabia, overseas market authorisation evidence plays a supportive but not standalone role in the medical device and IVD registration process. The regulatory authority is the Saudi Food and Drug Authority (SFDA8), and all devices must obtain Medical Device Marketing Authorization before being marketed. The CE certificate is typically part of the supportive information, but it does not replace the need for full documentation and conformity assessment.
Serbia
Serbia is located in Europe but is not part of the EU or EEA. In Serbia, the use of overseas market authorisation evidence is recognized and can support the registration of medical devices and IVDs, but it is not sufficient on its own. All devices must be registered with the Medicines and Medical Devices Agency of Serbia (ALIMS9) before being placed on the market. Serbia does not have a formal regulatory reliance pathway, but it does accept documentation from recognized jurisdictions (including the EU) to support the evaluation process. Serbia is aligning with EU standards as part of its EU accession process. The CE mark is accepted for medical devices if the product complies with Serbian technical regulations.
Singapore
In Singapore, the Health Sciences Authority (HSA10) allows the use of overseas market authorisation evidence to support the registration of medical devices and IVDs, particularly through its abridged evaluation routes. This is part of Singapore’s risk-based and internationally aligned regulatory framework. Singapore recognizes approvals from several reference regulatory agencies, including EU notified bodies. If a device has been CE marked, it may qualify for abridged evaluation in Singapore.
Switzerland
Switzerland is located in Europe but is not part of the EU or EEA. In Switzerland, the use of overseas market authorisation evidence, especially CE marking, has traditionally been central to medical device regulation. However, due to changes in the EU-Switzerland Mutual Recognition Agreement (MRA), the regulatory landscape has evolved, and Switzerland is now adapting its approach to include controlled acceptance of non-EU approvals, such as those from the US FDA. Switzerland primarily relies on CE marking but is now planning to open up to FDA-cleared devices through a controlled pathway. This marks a shift toward regulatory diversification, while still maintaining high safety and performance standards. Swissmedic11 is the national authority responsible for medical device regulation.
Turkey (Türkiye)
Turkey is partially located in Europe but is not part of the EU or EEA. In Turkey, overseas market authorisation evidence, especially CE marking, is central to the medical device regulatory framework due to the EU–Turkey Customs Union. Turkey has fully harmonized its medical device regulations with the MDR and IVDR. Medical devices and IVDs must bear the CE mark to be placed on the Turkish market. CE marking is accepted as proof of conformity with Turkish regulations, which are aligned with EU directives. The Turkish Medicines and Medical Devices Agency (TMMDA12) is the national authority responsible for device regulation.
United Kingdom
In the United Kingdom, the use of overseas market authorisation evidence for medical devices and IVDs is evolving under the post-Brexit regulatory framework. The UK now operates a distinct system for Great Britain (England, Scotland, and Wales) and Northern Ireland, with different rules for each. CE-marked medical devices and IVDs can be placed on the GB market until 30 June 2030. After that, UKCA marking becomes mandatory*, unless the proposed new international reliance routes are formally adopted. The Medicines and Healthcare products Regulatory Agency (MHRA13) is the national authority responsible for device regulation.
* Will it, indeed? The MHRA have just announced that they will open further consultation on whether to recognise EU CE marked medical devices indefinitely due to the feedback received during the previous consultation on international reliance schemes. Whilst the MHRA’s recent response to that previous consultation also indicates that they are going to move forward with a reliance scheme that accommodates ‘approvals’ from comparable regulator countries (Australia, Canada and the USA)14.
Summary
While the CE mark is primarily for the EU and EEA, many countries outside the EU/EEA recognize or rely on the CE mark to streamline their own regulatory processes. Several non-EU/EEA countries accept or rely on CE marking to varying degrees and for some it is mandatory. This reliance and recognition can be unilateral (one-way acceptance) or mutual (based on formal agreements) such as mutual recognition agreements. Manufacturers who make use of the CE mark in this way also need to be aware of any national legislation in those countries. While the CE mark may open the door, national legislation may cover things like language requirements, unique device identification (UDI), and postmarket surveillance.
Related next steps
If you’re having difficulty navigating the EU regulatory landscape and struggling to comply with horizontal EU regulations, our experts specialise in helping manufacturers develop comprehensive regulatory strategies designed to ensure that medical devices and IVDs comply with all applicable EU legislation.
Contact us to discuss how we can support your efforts and stay tuned for more updates related to the European Medical Device landscape in our upcoming blog posts.
- https://akbpm.gov.al/
- https://www.tga.gov.au/
- https://almbih.gov.ba/en/agencija-za-lijekove-i-medicinska-sredstva-bosne-i-hercegovine-eng/
- Medical Device Division, Ministry of Health
- https://www.mda.gov.my/
- https://cinmed.me/en/
- https://malmed.gov.mk/
- https://www.sfda.gov.sa/en
- https://www.alims.gov.rs/english/
- https://www.hsa.gov.sg/
- https://www.swissmedic.ch/swissmedic/de/home.html
- https://www.titck.gov.tr/
- https://www.gov.uk/government/organisations/medicines-and-healthcare-products-regulatory-agency
- https://www.gov.uk/government/consultations/consultation-on-medical-devices-regulations-routes-to-market-and-in-vitro-diagnostic-devices/outcome/response-to-international-reliance-ukca-marking-and-in-vitro-diagnostic-devices-consultation-proposal