




By Gee Burke MD PhD, recently former FDA (CDER)
Ask any manufacturer of combination products, and they’ll tell you the same thing: one wrong move with the FDA’s three-headed monster: CDER, CBER, and CDRH, and your program can vanish into regulatory purgatory. I know, because I used to sit on the inside of that labyrinth at FDA. Combination products promise enormous clinical impact. But if you don’t know how to navigate the Bermuda Triangle formed by CDER, CBER, and CDRH, you may be sailing blind while you incorrectly assume you are charting a path to approval.
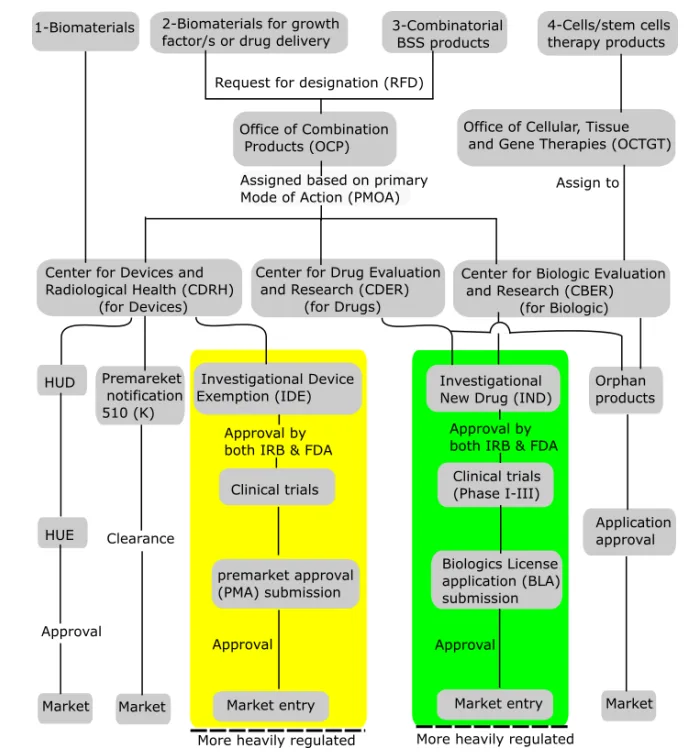
Who Really Runs the Show? Understanding the Centers
The FDA regulates combination products through three key centers:
- CDER (Center for Drug Evaluation and Research) for drug-led products.
- CBER (Center for Biologics Evaluation and Research) for biologic-led products.
- CDRH (Center for Devices and Radiological Health) for device-led products.
The lead depends on the product’s Primary Mode of Action (PMOA), the component of the product that provides the principal therapeutic effect. If the PMOA is unclear, the FDA Office of Combination Products (OCP) can assign the responsible center via a Request for Designation (RFD) process.
Insider reality check
In practice, this “assignment” isn’t always as neat as the flowcharts make it look. Centers often jockey behind the scenes, and if you haven’t prepared your justification carefully, expect delays, or worse, conflicting asks from multiple reviewers.
Real-World Examples (and What They Teach Us)
- Drug-Eluting Stents: Typically reviewed by CDRH, but CDER’s input on drug kinetics is critical. I’ve seen too many teams stumble here by underestimating how early CDER wants to see stability data. Don’t make that mistake.
- Prefilled Syringes:. Syringes with vaccines, insulin, or biologics sit under CDER or CBER, with CDRH chiming in. What people forget: human factors data can sink you if you treat the syringe like “just a container.”
- Transdermal Patches: This device/drug combination delivers medication through the skin. CDER commonly leads with advice from CDRH. The trap? Oversimplifying device manufacturing controls. FDA won’t.
- Encapsulated Cellular Therapies:. CBER usually leads, but device performance is central. Here, reviewers ask questions most teams never anticipate, like device biocompatibility over long-term implantation.
The Regulatory Bermuda Triangle in Action
Jurisdictional Complexity
Combination products demand careful center assignment, as the lead center sets the regulatory requirements (GMP, QSR, IND, NDA, BLA, 510(k), PMA, etc.) and post-market obligations. Ambiguous PMOAs can generate jurisdictional disputes, risking costly review delays.
Intercenter Collaboration
The reviewing center often lacks deep expertise in all product aspects. Intercenter agreements allow consultation (e.g., CBER might lead but must seek CDRH advice for a device component). Proactive, early communication with FDA and clear documentation about product attributes help streamline assignments and avoid confusion. Inside FDA, I watched files stall simply because one center hadn’t responded to another’s consult request.
Regulatory Pathway Cheat Sheet
|
Product Example |
Components |
Lead Center Designation |
Submission Type |
|---|---|---|---|
|
Drug-eluting stent |
Device & Drug |
CDRH or CDER |
PMA & NDA combo |
|
Prefilled syringe |
Device & Drug/Biologic |
CDER or CBER |
NDA/BLA & device data |
|
Transdermal patch |
Device & Drug |
CDER |
NDA, with device data |
|
Cell therapy w/ device |
Device & Biologic |
CBER |
BLA & device section |
Pro tip
Don’t treat this table as gospel. FDA may flex based on evolving science, safety signals, or political pressures. Always verify assumptions early.
Surviving the Triangle: Practical Tips
- Map your product’s modes of action early and prepare justification for PMOA.
- Engage FDA through Pre-RFDs or formal RFDs at the earliest opportunity.
- Build integrated development plans that cover drug, device, and biologic regulations (e.g., 21 CFR 210/211 and 820, as needed).
- Expect and plan for cross-center consultation. No single center holds all the expertise your product may require for approval.
- Maintain regulatory updates in FDA policies or OCP guidance.
Emerging watchpoints: Digital drug-delivery platforms, AI-enabled injectors, and gene/cell therapies with embedded delivery devices are rewriting the rules as we speak. If your regulatory strategy looks backward instead of forward, you’re already behind.
Final Word
Combination products open new therapeutic frontiers, but their regulatory path requires focus to avoid getting lost in the Bermuda Triangle. Mastering center assignments, regulatory pathways, and necessary cross-functional collaboration is the route to both regulatory survival and commercial success. Combination products don’t have to disappear into the regulatory abyss. With the right strategy, you can chart a safe course. Having lived inside the FDA’s Bermuda Triangle, team RQM+ with expert strategic and tactical guidance from me help manufacturers be a regulatory and commercial success.
If your program sits at the intersection of CDER, CBER, and CDRH, don’t leave your future to chance. This is where my experience inside the FDA becomes your greatest competitive edge.
Exploring MedTech Consulting?
Full-service support isn’t just about doing more, it’s about designing stronger, more resilient product strategies from day one. Whether you’re bringing a first device to U.S. market or responding to shifting regulations, a partner like RQM+ brings the agility, depth, and cross-functional horsepower to accelerate progress with confidence. For MedTech leaders, the upside is clear: fewer delays, lower risk, and a faster path to clinical impact.
Prefer to start quickly? Begin with a consultative conversation.
- Naderi-Meshkin, H et al. (2018). Critical Issues in Successful Production of Skin Substitutes for Wound Healing. Journal of Genes and Cells. 4. 10. 10.15562/gnc.63.